Electronic Structure Theory
Knowledge of the electronic energy and the way electrons assemble in molecules and matter underpins our understanding of chemical structure, reactivity, dynamics and light-matter interactions. We develop sophisticated computational methods to accurately solve the electronic Schrödinger equation numerically for complex systems.
Accurate, efficient and robust ab initio electronic structure methods
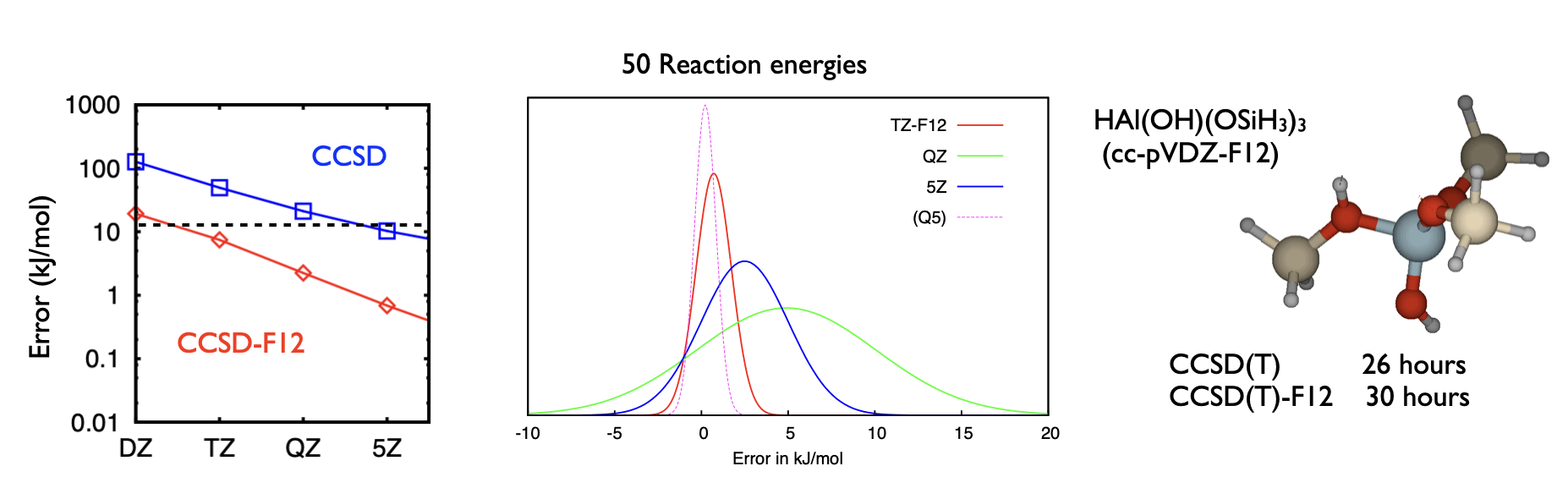
Accurate ab initio prediction of electronic energies requires a sufficiently complete treatment of electron correlation. For most molecules, this means accounting for at least two- and three-body electron correlation processes and correlation-induced orbital relaxation in the wavefunction form. In addition, the parameterisation of these correlation processes must be sufficiently flexible to capture the finer details of the correlation function, which means in practice using large atomic basis sets (at least QZ quality).
We develop explicitly correlated wavefunction theory. This approach introduces explicit functions of inter-electron separation into the wavefunction parameterisation, properly modelling the derivative discontinuities that arise in the wavefunction whenever two electrons meet. This F12 methodology makes it possible to obtain accurate electronic energies with small, DFT-sized basis sets, resulting in a computational saving of two orders of magnitude over standard approaches of comparable accuracy.
Low-scaling correlation treatments for periodic Systems
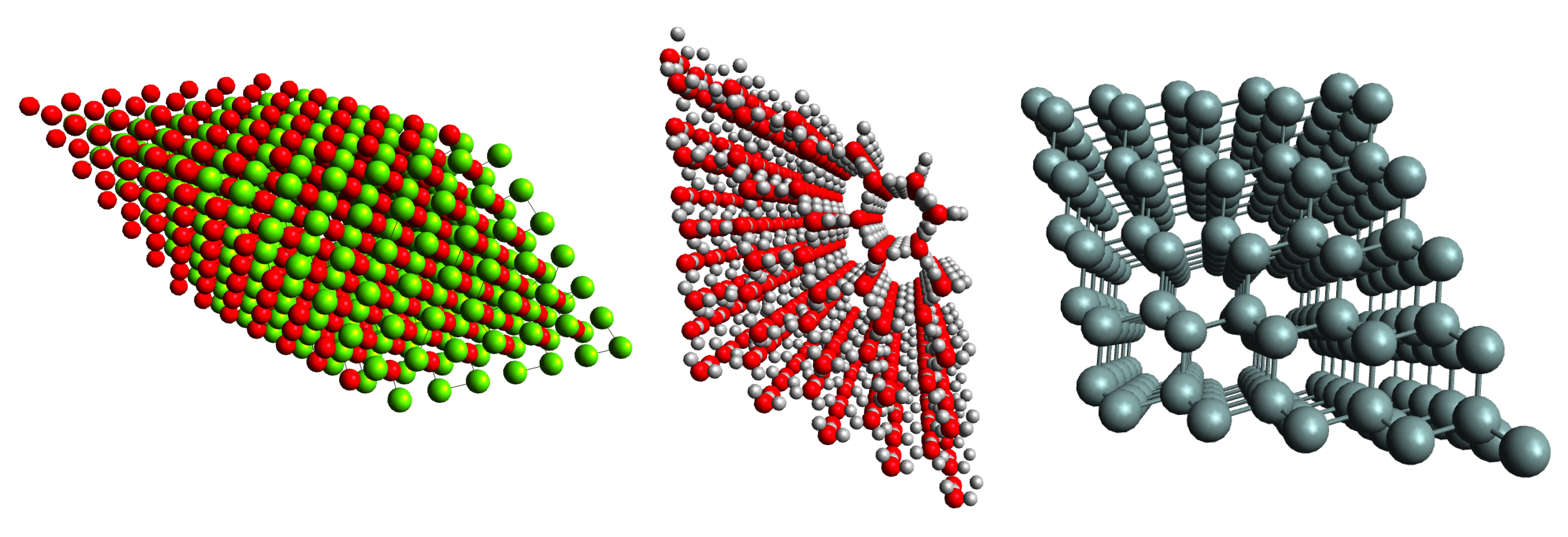
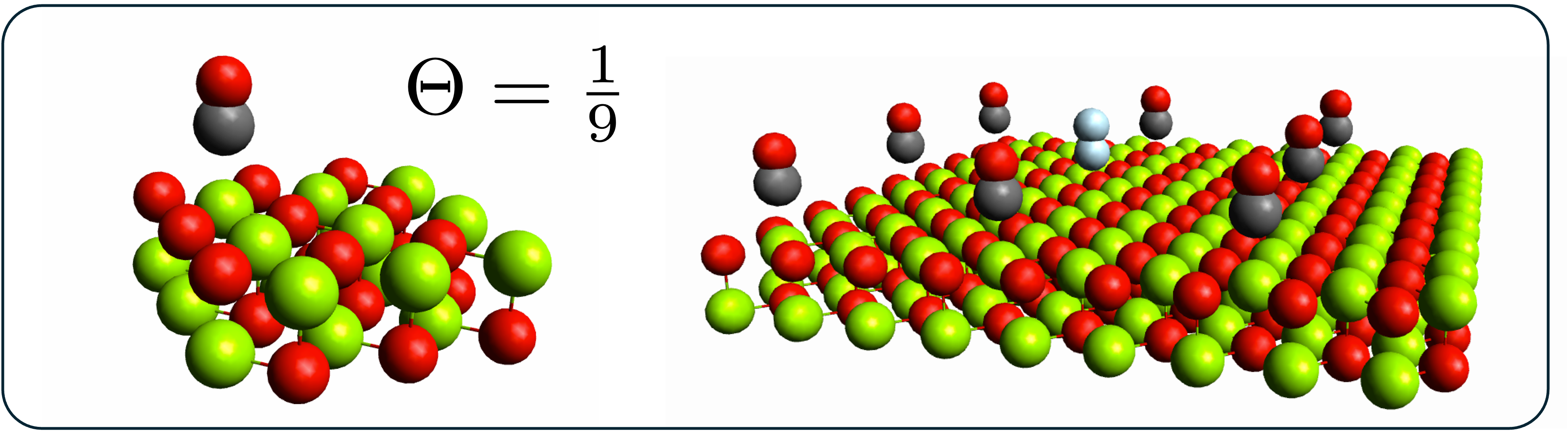
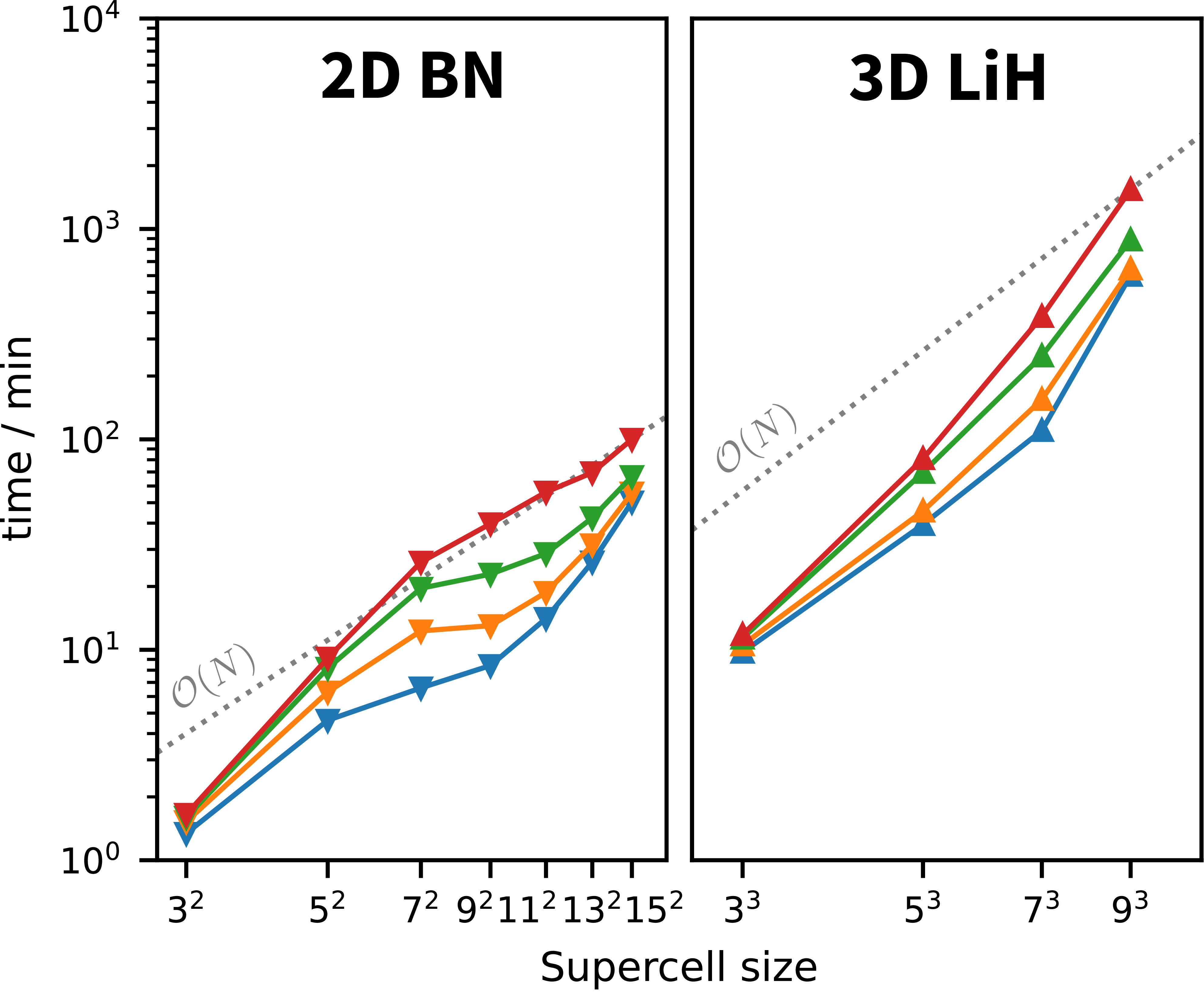
Accurate correlated wavefunction methods for periodic systems remain computationally demanding as either the unit-cell size or the number of simulated cells increases. In their conventional canonical formulations, methods such as MP2 and CCSD(T) scale as \(n^5\) and \(n^7\), respectively, which limits their application to realistic materials problems.
Pair natural orbital (PNO) methods provide a well-established route to reducing this cost in molecular electronic structure theory. By exploiting the locality of electronic correlation, they construct accurate low-rank approximations to excitation amplitudes and electron repulsion integrals. Our work extends these ideas to systems with periodic boundary conditions, with the goal of developing linear- or near-linear-scaling algorithms for solid-state and materials applications.
These methods are implemented directly within the TURBOMOLE package. This enables us to study ground-state properties such as adsorption energies, lattice constants, and formation energies for systems of a size that approaches the practical reach of density functional theory, while delivering the improved accuracy of correlated wavefunction approaches.
Multi-reference treatments
Coupled cluster (CC) theory based on a single Slater determinant is a cornerstone of high-accuracy electronic structure theory. However, many chemically important systems, including radicals, transition-metal complexes, and molecules far from equilibrium geometries, cannot be described adequately from a single-reference picture. In these cases, a multi-determinantal reference is needed to capture strong or static correlation.
This distinction between static and dynamic correlation has shaped the development of quantum chemical methods for decades. Configuration interaction (CI), with its linear ansatz, is naturally suited to describing static correlation, but it does not provide dynamic correlation in a size-consistent and size-extensive way. By contrast, methods such as many-body perturbation theory (MBPT) and coupled cluster theory employ wavefunction forms with the correct scaling behaviour, making them better suited to the treatment of dynamic correlation.
The development of multi-reference coupled cluster (MRCC) methods that capture both static and dynamic correlation in the most general setting remains a major challenge. In many important cases, however, symmetries such as spin define a single configuration state function (CSF) as a suitable reference, so that the remaining problem is one of adding dynamic correlation to an already appropriate zeroth-order state.
In our group, we develop coupled cluster approaches for such multi-determinantal reference states. Our work builds on Lindgren’s idea of using a normal-ordered exponential (NOE) ansatz, which simplifies the coupled cluster working equations while retaining the desirable structure of the theory. By combining this framework with modern equation-generation techniques, we have implemented practical methods and applied them to a range of strongly correlated molecular systems.
More recently, we have developed a generalised normal-ordered coupled cluster (GNOCC) framework, in which the normal ordering of the NOE is defined with respect to the correlated reference state itself. This allows us to treat arbitrary spin eigenfunctions, including configuration state functions and CASSCF wavefunctions, within a coupled cluster formulation. As a result, we can perform state-specific, spin-free coupled cluster calculations for both ground and excited states in a manner that is size-extensive and size-consistent.