Quantum Computing for Chemistry
State Preparation
Preparing a good initial guess is often the limiting step in quantum algorithms for chemistry: the closer the starting state is to the true eigenstate, the shallower the circuits and the fewer shots are needed downstream. Our work in this area focuses on encoding chemical structure, particularly spin symmetry and electron correlation, directly into compact, shallow circuits, rather than relying on generic heuristics.
Papers
- Marti-Dafcik, Burton, Tew. Spin coupling is all you need: Encoding strong electron correlation in molecules on quantum computers. Phys. Rev. Research 7, 013191 (2025) — deterministic O(N)-depth circuits preparing spin-coupled initial states with large overlap on strongly correlated targets.
- Marti-Dafcik, Lee, Burton, Tew. Spin-coupled molecular orbitals: chemical intuition meets quantum chemistry. arXiv:2402.08858 (2024) — revives spin-coupled MO theory as a physically motivated ansatz class for diradicals and bond-breaking.
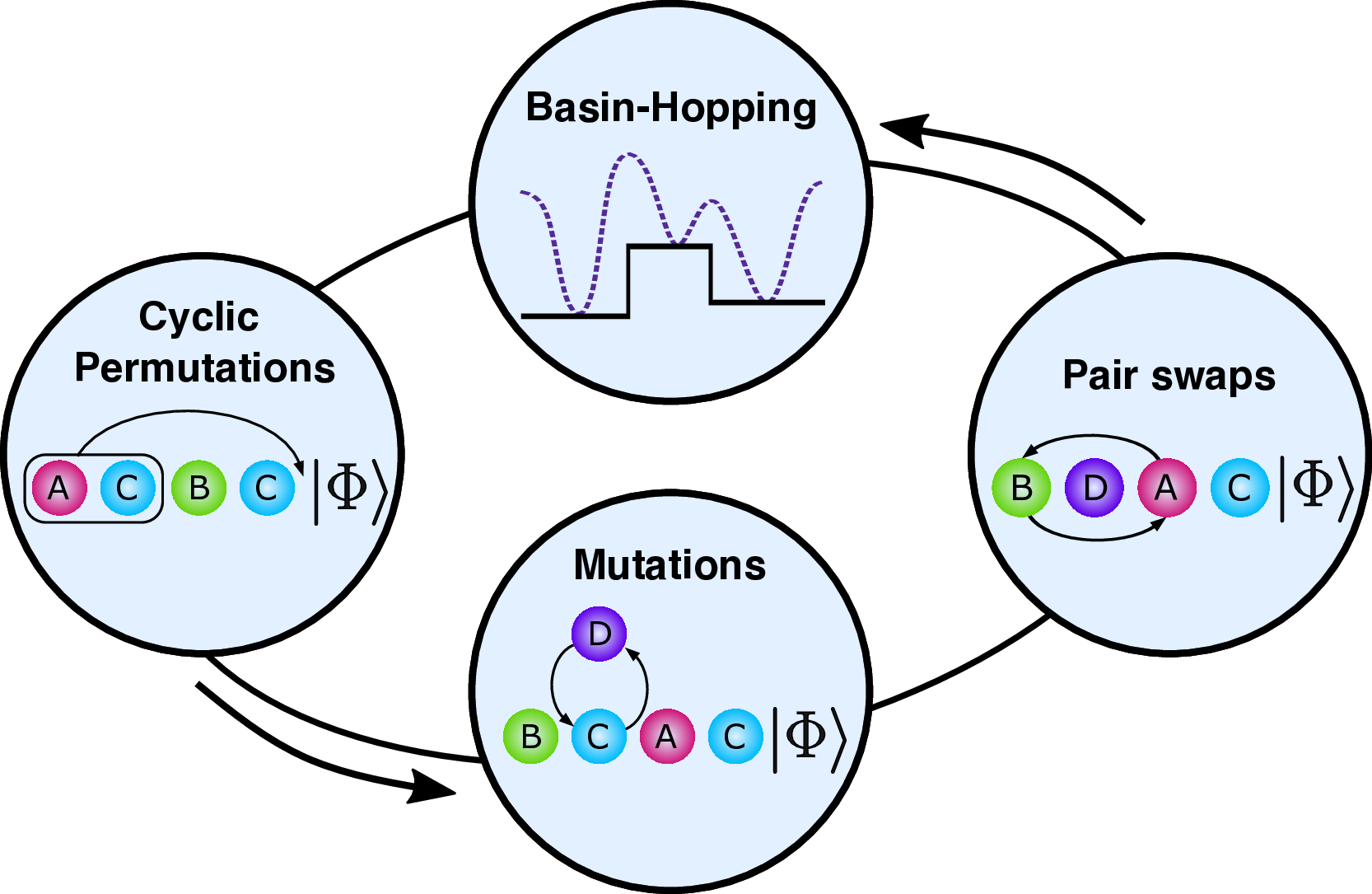
- Burton, Marti-Dafcik, Tew, Wales. Exact electronic states with shallow quantum circuits from global optimisation. npj Quantum Information 9, 75 (2023) — jointly optimises ansatz structure and parameters to maximise accuracy at fixed circuit depth.
Simulating Dynamics
Time evolution on quantum hardware is a natural route to absorption spectra, photodynamics and electron dynamics that are hard for classical methods. We work on grid-based (first-quantised) algorithms for wavepacket propagation and on measurement-light protocols that extract spectral information from short simulations, with careful end-to-end resource analysis for realistic molecules.
Papers
- Feng, Chan, Tew. Quantum Resource Assay for the Grid-Based Simulation of the Photodynamics of Pyrazine. arXiv:2506.08609 (2025) — full fault-tolerant resource estimate for absorption spectra and vibronic population dynamics of pyrazine (17 → 97 qubits across 2D to 24-mode models).
- Chan, Meister, Jones, Tew, Benjamin. Grid-based methods for chemistry simulations on a quantum computer. Science Advances 9, eabo7484 (2023) — split-operator QFT methods for 2D/3D atoms, covering ground-state preparation, ionisation and scattering dynamics on up to 36-qubit emulations.
- Chan, Meister, Goh, Koczor, Tew (co-author). Algorithmic Shadow Spectroscopy. PRX Quantum 6, 010352 (2025) — extracts Hamiltonian energy gaps from time-evolved classical shadows with minimal shot cost, demonstrated on IBM hardware up to 100-qubit emulations.
Quantum Eigensolvers
Eigenvalue estimation of ground- and excited-state energies is the flagship chemistry application of quantum computers. In the past, our work has bridged classical electronic-structure theory with VQE-style and projective eigensolvers, targeting ansätze that respect molecular symmetries, handle strong correlation, and reach excited states at reduced circuit depth. We are currently looking at algorithms that leverage quantum advantage to avoid reliance on chemical intuition.
Papers
- Chan, Fitzpatrick, Segarra-Martí, Bearpark, Tew. Molecular excited state calculations with adaptive wavefunctions on a quantum eigensolver emulation: reducing circuit depth and separating spin states. Phys. Chem. Chem. Phys. 23, 26438 (2021) — adaptive Variational Quantum Deflation for excited states with spin-state resolution.
- Burton, Marti-Dafcik, Tew, Wales. Exact electronic states with shallow quantum circuits from global optimisation. npj Quantum Information 9, 75 (2023) — also a central eigensolver-ansatz-design contribution (cross-listed with Theme 1).
- Marti-Dafcik, Burton, Tew. Spin coupling is all you need. Phys. Rev. Research 7, 013191 (2025) — though framed as state preparation, its motivation is improving overlap for eigenvalue algorithms (QPE, VQE), so also fits here (cross-listed with Theme 1).